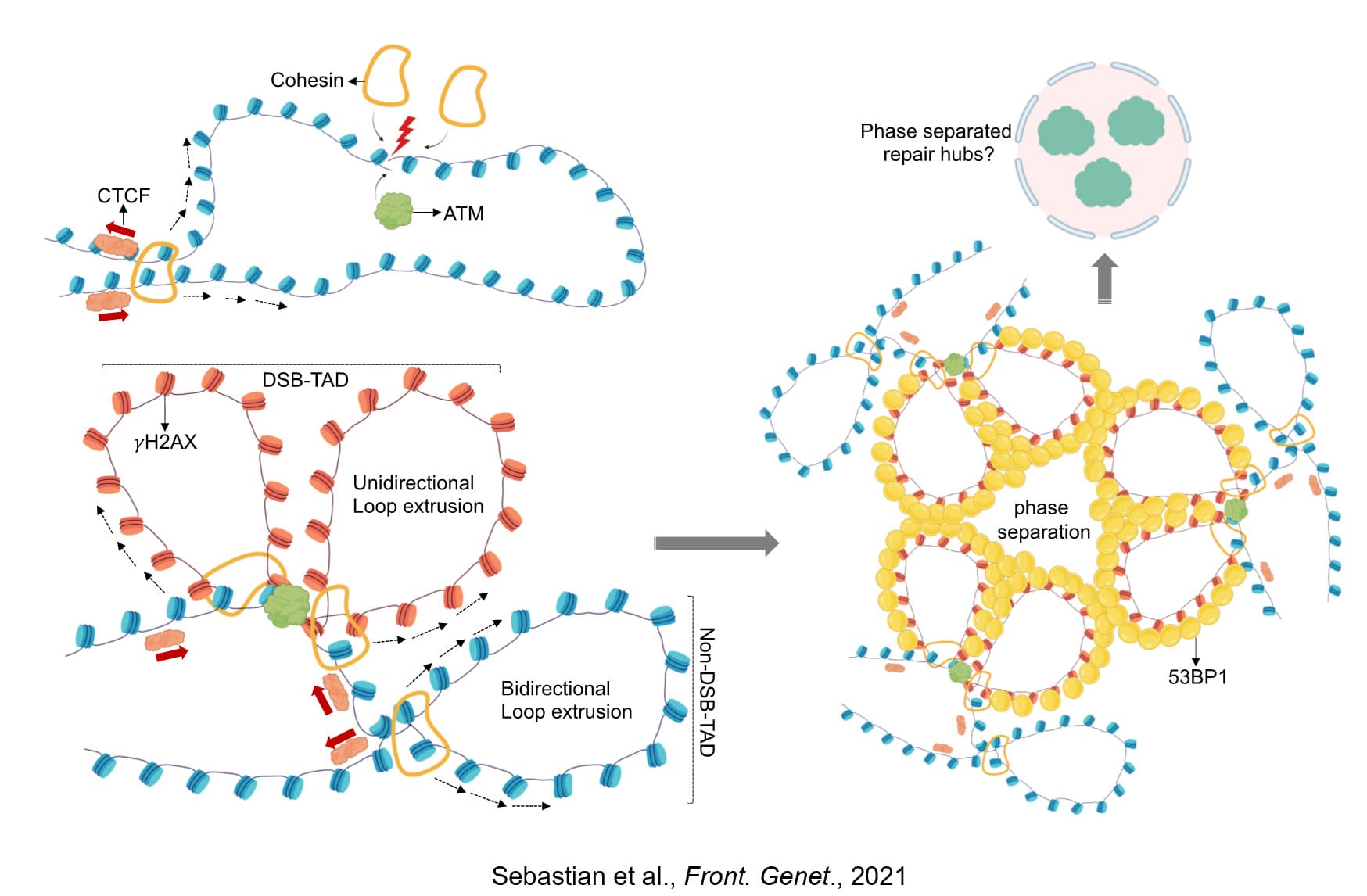
Mechanisms of replication origin initiation control in response to DSB induction
Using DSBs as a model of genotoxic insult, we discovered that the initial response of DNA replication to DSBs is more local than global (Sebastian et al., Nature, 2025). We found that DSBs induce a local genome maintenance mechanism that inhibits replication initiation in DSB-containing topologically associating domains (TADs––the fundamental structures of genome architecture) without affecting DNA synthesis at other genomic locations. This gives cells an opportunity to respond to the insult yet continue replication elsewhere. We named this mechanism "response of Mediators of Replication and DSBs" or "MRD response". Using a high-throughput RNAi screen, we then identified MRD factors that regulate this response. This discovery demonstrates that genome stability is intricately linked to genome architecture and spatiotemporal regulation of mammalian DNA replication. We further found that licensed replication origins at the vicinity of DSBs are not activated. Our lab is interested in deciphering the biochemical mechanism that selectively suppress replication initiation from DSB-proximal replication origins.
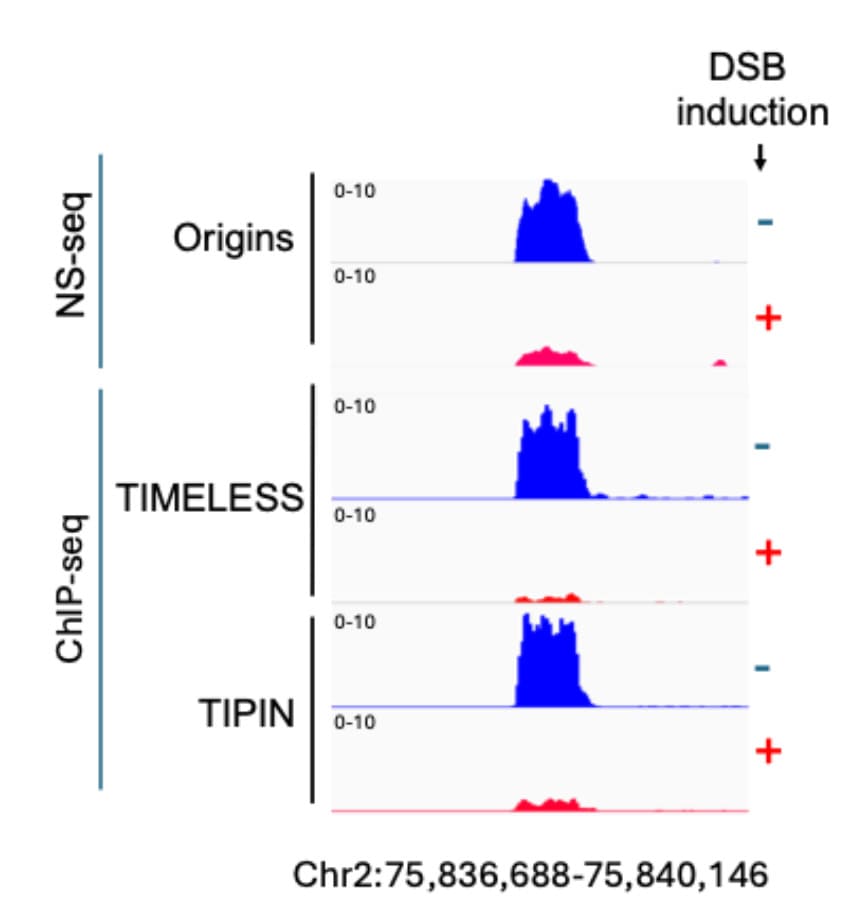
MRD factors TIMELESS and TIPIN are dissociated from replication origins that are suppressed after DSB induction.